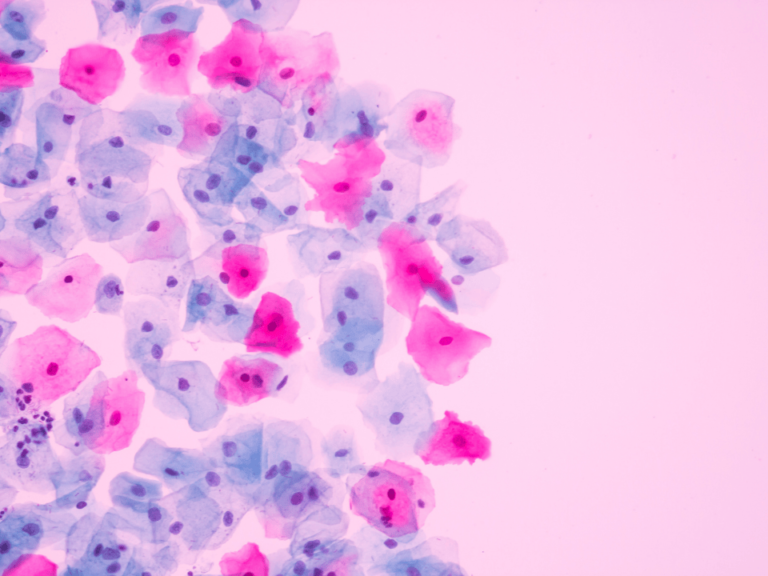
FDA’s Class II 510(k) clearance process for medium-risk devices—a category that includes the power morcellator—is inadequate, because it does not focus on risk assessment, according to Bill Vodra, a former associate chief counsel for drugs at FDA.
Instead, the 510(k) process relies on “substantial equivalence” to predicate devices, thereby allowing subsequent iterations of a device to introduce risk without active FDA surveillance.
“A huge variety of devices are now in Class II, and they pose extraordinarily different kinds of risk,” Vodra said. “The current test for clearance of a 510(k) is, ‘Is the proposed device substantially equivalent to another device (the predicate device or device chain) that has been marketed?’
“The answer may be yes, but that does not tell you much about risk of the proposed device or its predicates.”
Vodra, a retired partner of Washington, D.C. law firm Arnold and Porter, helped draft many FDA regulations still in use, including those implementing the Controlled Substances Act and FDA’s rules for Good Manufacturing Practices, Good Laboratory Practices, Good Clinical Practices, bioequivalency and the Orange Book.
“You start by having a question in the preclearance process: what do we know about the risk posed by this device and its predicates?” Vodra said. “That could mean breaking apart the current Class II devices into a much larger universe of disparate device groups, because different groups of devices present risks similar to each other, but distinct from other groups of devices.”
Vodra spoke with Matthew Ong, a reporter with The Cancer Letter.
Matthew Ong: What is the difference between how drugs and devices should regulated? What are the primary reasons for why drugs and devices have different pathways for regulation?
Bill Vodra: With all medical interventions, the goal is to assure that they provide a reasonable assurance that they will deliver the benefit they promise (i.e., are effective), and that the risks they present are outweighed by these benefits (i.e., are safe). But how one develops the evidence of safety and effectiveness differs markedly between drugs and devices.
Fundamentally, I would call it a matter of n, the numbers of humans on which you can test a new technology in a reasonable period of time.
With drugs, the process for developing a drug starts with an identified and well-characterized molecule—sometimes with biologics that’s not quite true—but normally, you know exactly what it looks like, and you analyze it in a laboratory.
You can control the dosage of drugs very specifically, and then you can give that in a staged fashion, first to normal subjects. These are people who are not sick in any way, shape or form, and you’re looking for the things that animals can’t tell you—rats and mice don’t talk to you about headaches, dizziness or nausea.
You’re really looking for safety issues. There are things you learn in phase I studies working the dosage up until you’re well into the range where you’re expecting therapeutic effects.
Then you go to people with the disease you are trying to study in phase II trials and that you’re trying to get this drug to work in. You start with a small number of patients, and monitoring them extremely closely, often in the clinical setting, to determine if these patients react differently than normal subjects would.
Once you establish the doses, you then take the drug into larger populations, essentially the phase III studies, often with 3,000 to 5,000 people in them. And now we’re into Bayesian statistics, adaptive design trials and so forth, where you look at who responds and who doesn’t respond, and ask why. Studies can be modified or tailored to the patients who predictably should respond.
That’s the drug model, and it relies on the availability of a large number of people in whom to test the drug in a reasonably fast manner.
Go to the device situation: generally, the number of available patients in a given period of time, and the existence of a thoroughly standardized product throughout that period, are radically different.
You would use animals first, but obviously, when you finally get to humans, you don’t go to phase I normal subjects. You go right to phase II, and when you find a device doesn’t work, you go back and tinker with the product and make changes.
You may have many generations of products out there before you settle on the one you want to launch in the marketplace, and then you learn from the marketplace that you need more modifications. Look how frequently you get Microsoft updates—that’s very typical of what they’re doing in all engineering areas. The device continues to evolve in a way that a drug does not.
The model for drugs simply does not work for devices. So we have developed an alternative system in which a few high-risk devices are reviewed by FDA via a premarket application with extensive safety and effectiveness data, others are reviewed by a 510(k) submission with limited clinical data, others are reviewed by a 510(k) submission without clinical data, and even others are not reviewed at all before entering the marketplace.
Can you explain the 510(k) process we have today?
BV: When the law was being drafted in 1976, it originally had only two classes—one was going to be preclearance through the premarket approval process (the current Class III), and one was going to be essentially without any review whatsoever (the current Class I). Because of that, there was a concern that FDA would put everything into the preclearance mode, which was going to break the whole system down and burden many devices unnecessarily.
So the drafters came up with this intermediate Class II system, under which access to the market would be by showing conformity to a set of regulatory standards (e.g., diagnostic sensitivity and specificity, or wavelength and focal point size of a therapeutic laser). In the interim, pending development and promulgation of regulatory standards for Class II devices, and requirements for PMAs for Class III devices, a “temporary” mechanism was established, under which new devices could enter the market by demonstrating “substantial equivalence” to a device on the market in 1976. That then morphed over time to become a permanent feature for Class II devices.
In practice, the assumption that if the risk was acceptable to the previous devices, it is acceptable for this device, does not even ask, “Have we made any steps to reduce that risk?” It only asks whether the risk any worse with the new product.
And if the answer is not obviously “yes,” the product gets cleared. That makes no sense to me. That’s not focusing on the risks actually posed and attempting to manage or reduce those risks.
This story is laid out very clearly in the appendix to the IOM report in 2011. Both FDA and the industry are wedded to the “substantial equivalence” standard, which is perceived to be something that was far less demanding for gaining marketing access for Class II products than the premarket application process required of Class III devices.
We’ve talked in the past about whether the 510(k) clearance process adequately protects patients from harm. Advocates are now saying that a more reliable risk-based evaluation system needs to be instituted. What could that be? Is it possible to come up with something that’s better than what we have?
BV: The short answer is, yes, we can do more than we’re doing. The current test for clearance of a 510(k) is, “Is the proposed device substantially equivalent to another device (the predicate device or device chain) that has been marketed?” The answer may be yes, but that does not tell you much about risk of the proposed device or its predicates.
The implicit assumption of the substantial equivalence standard for the 510(k) is: “We’ve lived with that earlier device; therefore we can live with this one.” In fact, we know from a number of case studies that the risks of the predicate device (or devices) might never have been identified or understood.
For a comprehensive evaluation of the 510(k) process, see the Institute of Medicine report in 2011. The whole premise of the IOM criticism was, if FDA doesn’t ask about the risks, it’s not necessarily going to get answers about risk.
For specific proposals on how to look into existing databases that the agency or the industry to determine whether or not risks have been identified properly in the past, refer to a FDLI Food and Drug Policy Forum that I wrote later that year. I note that manufacturers and FDA have a variety of historic records that can be explored to look at this question.
What is your fix for this? How do you actively and proactively assess risk?
BV: You start by having a question in the preclearance process: what do we know about the risk posed by this device and its predicates? That could mean breaking apart the current Class II devices into a much larger universe of disparate device groups, because different groups of devices present risks similar to each other, but distinct from other groups of devices.
A huge variety of devices are now in Class II, and they pose extraordinarily different kinds of risk. Consider just diagnostic tools: you have in vitro diagnostics that do not come into contact with the human body, diagnostics that work by passively collecting information through contact with the body, diagnostics that work by emitting energy into the body, and in vivo diagnostics that are placed inside the body. All diagnostics must provide accurate information, of course, but beyond that, the different categories can raise unique risks not found in the other categories. The same analysis could be applied to implanted and external devices to affect heart rhythm, or contraceptive devices, or lasers for therapeutic use, and so on. We now have almost 40 years of regulating devices, which was not available in 1976. Surely we don’t need any longer to lump all of these disparate tools into a single “Class II” with a single standard of clearance, based fundamentally upon substantial equivalence to a device sold before 1977.
And yet, we don’t differentiate those devices and ask questions like, “What are the risks that we know about this, or that group of products, and what have we done to address those risks?”
Would this require more premarket testing of devices?
BV: Not necessarily. As I stated before, there is a fundamental difference between drugs and devices in terms of the number of patients you can study before you make a decision about proceeding to routine medical use. In drugs, you can get 5,000 patients, sometimes 10,000 patients, in premarket studies. In the device arena, it may be 300 to 500 patients or less.
If you’re looking for something that occurs in the incidence of one in 1,000—you’re not going to find it necessarily in the first 500 patients.
David Feigal, former CDRH director, has proposed what he called the “Lifecycle Iteration of Medical Devices” in which you use version 1.0 as the test model for version 2.0—find out what you can about version 1.0 through postmarket surveillance and experience, and then fix those things for version 2.0. Postmarket surveillance on version 2.0 tells you what changes you need to make for version 3.0, and so on.
What Feigal is suggesting is: Use the postmarket surveillance for the first-generation product, and then design the second-generation product.
With that, you’re studying what you know about this product, whereas currently, if version 2.0 is no riskier than version 1.0, it’ll go out on the market, and we don’t ask the question of, “Have we addressed the risks identified for version 1.0 and reduced those risks?”
How is the IOM 2011 proposal different from Feigal’s proposal?
BV: The IOM Committee was looking at a broader series of things than just that. Feigal was proposing how to use existing experience to improve devices, one device at a time.
We were looking at this systemically: What is the adverse outcomes reporting system like, and how can that be used to assure the safety of products in the marketplace, which is a little bit different, because it’s not, “How can you improve one particular product?” but simply, “How do you learn about what’s out there?”
For example, “How do you learn whether the morcellator is causing upstaging of cancer?” That was the kind of question we were focusing on. And we were saying that you can’t improve the 510(k) process, which was what we were charged with doing, without having a well-functioning postmarket surveillance system, which we don’t really have yet.
FDA wrote in a Nov. 12 letter to Rep. Mike Fitzpatrick (R-Pa.) that the agency disagrees with IOM’s recommendation about changing the 510(k) process. What is your take on that?
BV: FDA issued a statement like that the day the IOM published the report. The agency had received copies of the report a week prior to its public release, and had had a chance to consider it.
In fairness to everybody, I think that FDA did not expect what it got from our IOM panel. They were looking for a checklist of, “Tweak this, tweak that,” and not a “Throw the whole thing out” which, I fear, is how FDA first read the report. The IOM Committee said that FDA needs to fundamentally rethink it. There are places where the 510(k) process with “substantial equivalence” still makes a lot of sense.
I believe that FDA was taken totally off-guard and put in an awkward place. First, FDA certainly didn’t want the political problem of dealing with such a sweeping idea on the eve of having to renegotiate the user fee requirements for 2012. Industry was equally unwilling to address the idea, and legislative changes were just not politically possible. Secondly, what the IOM report basically said is that the 510(k) process is not doing as much to protect the public as the public perceived. That conclusion required a lot of people within the agency to swallow and say, “You mean everything we’ve done for the last 10 or 20 years is worthless?” It’s not quite the right interpretation of the IOM message, but that’s the way it could come across to the dedicated career staff at the agency.
At the end of it, FDA just said, “No, thank you.” But I’ve been told that there is an awful lot in the IOM report that FDA does agree with that and that it will seek to implement over time.
The IOM process itself is partially to blame. If we had been able to lay out a vision for what a new process or processes might look like, it might have been easier for FDA and industry to deal with it. As it happened, the committee was given a very strict timetable that could not be extended. By the point we had realized the fundamental deficiencies in the 510(k) system, we lacked the time to develop alternatives. My regret is that we didn’t have another year to come up with some additional suggestions. It would have been a lot easier for them to deal with. Plus, we could’ve gotten it out after the 2012 user fee negotiations, in which case FDA and industry would have had several years to think about it before the next round of device user fee legislation.
In short, to everyone’s misfortune, I fear the IOM put a dead fish on the table and FDA disposed of it quickly. But this is all speculation on my part.
Other proponents of the 510(k) process say that scrapping or changing it further would stifle medical device innovation.
BV: I’ve heard that cry for my entire career. “Regulation stifles innovation!”
There are two answers to this, both extremely relevant. First, regulation frequently stimulates innovation. The requirement for adequate and well-controlled clinical studies for approval of new drugs after 1962 led (after a period of adjustment lasting to the early 1980s) to three decades of enormous productivity in pharmaceuticals. This golden age was significantly due to the developments in the way drugs were developed to meet the new regulatory standard. Secondly, innovation is not always inevitably beneficial. When you look at products that had gotten into the market that injured or killed patients, you have to acknowledge that innovation is a risky exercise.
In my experience, there are not many things that aren’t improved by having a second set of independent eyes looking at it. Whether it’s planning to invade Normandy in 1944 or to launch a new artificial heart, having somebody else look at the plans and ask questions is a very constructive process.
You’re saying that this argument that regulation stifles innovation has a long beard.
BV: Yes. To prove that argument, you have to show where in the world we’ve got more medical progress with less regulation.
In the 1970s, we had a debate called “drug lag,” which was an argument that new drugs were getting on the market much sooner in Europe than in the U.S., because the 1962 law had toughened things up, and took far more time to get new drugs through FDA than elsewhere. The advocates held up some examples of products, and FDA challenged them. I’m not sure either FDA or the drug lag advocates prevailed based on the experience of the 1970s. But by the end of the 1980s, the debate had evaporated, as FDA demonstrated consistently shorter review times and more drug approvals than all other advanced nations.
I’d like to see a similar evidence-based debate over the effect of FDA regulation on the development and entry of new devices to the U.S. and foreign markets, and the medical costs and benefits of the devices.
The ideology that regulation destroys innovation—I’ve been through it, and I’ve thought about it, and it doesn’t persuade me as more than rhetoric and speculation.
Let’s talk about the federal mandate for adverse outcomes reporting. Does the system work? Is it effective?
BV: Getting someone to report an adverse outcome to a manufacturer or FDA is about step 3 or 4 in a multi-step process.
First, you have to have somebody who has an adverse outcome and recognizes an adverse outcome. Usually, that means an outcome that is not expected with the disease. If you’re dealing with a drug or device intended to prevent heart attack, and the patient dies of heart attack, you don’t necessarily focus on, “Could the drug or device have caused the heart attack?” People don’t tend even to recognize that there is something unusual, if it seems part of the disease being treated.
The second step is, somebody has got to recognize that, not only is the outcome is untoward, but that it might also be associated with the exposure to some sort of intervention—a drug or device. So if the patient has a heart attack but you weren’t expecting that they would, you’ve got to say, “Could it have been the pacemaker? Or could it have been a drug he’s taking?” To connect the dots and say, “Gee, I wonder if that could’ve been a relationship”—somebody at the frontline, usually a patient, doctor or caregiver, has to make that association.
Usually, if there is a temporal relationship, like immediately after taking a drug, it’s more obvious. Where you’ve got something implanted in the body for a long time, it may be a lot harder to link the event with the device. That recognition may require seeing the same event in several patients.
Once somebody makes that association—that, maybe this intervention is related to that untoward outcome—then they have to be motivated to report it. Our entire system relates to voluntary reporting. We don’t have, with the exception of some user reporting requirements, a mandatory reporting system, in which everybody reports everything that happens to the patients. Some people argue that fear of malpractice liability inhibits voluntary reporting by physicians and hospitals; and others contend that plaintiffs’ attorneys in the area of product liability stimulate inappropriate and inaccurate reporting. So you have biases that can influence the frequency and quality of voluntary reports.
The next step is to get the report to someone who is responsible for collecting and investigating such reports. In America, there are three bodies that are charged with doing this: the manufacturers of the products, the FDA, and private registries that track certain types of devices. But reports may die in the pathway to these bodies. Patients tell doctors, who decide not to report; doctors tell hospitals, who decided not to report; and reports can simply go astray.
Once somebody voluntary reports an untoward event and its possible association to the manufacturer of the device, however, the company is legally required to investigate that report and determine whether it meets criteria for reporting forward to the FDA. Not every event is required to be reported to the agency.
The company is also charged with looking for patterns. It has been said that one case is an accident, two cases are a coincidence, but three cases suggests a pattern. Looking for patterns is a pretty sophisticated science, under the heading of epidemiology. The pharmaceutical industry has whole departments under the heading “pharmacoepidemiology.” Unless the device industry has changed radically in the last three or four years, my experience is that smaller device companies frequently don’t even have an epidemiologist on staff or on call.
So this sequence must all to fall into place in order to get that light bulb going off that says, “Yes, we’ve got a probable causal relationship here, or at least an association that is worthy of study.”
Individual companies have a further challenge, in that they follow only their own products. Absent publicly available registries or use of FDA’s public databases on medical device reports, it is impossible for a company to know whether the reports about its products are comparable to, or out of line with, those of competing products.
In summary, you’re saying that it’s difficult to capture adverse outcomes with voluntary reporting.
BV: Extremely difficult to identify, capture, investigate and interpret. That’s why I’m not so judgmental, off the top of my head, that a company should or should not have reported a medical problem until I know the facts of a particular case.
What about power morcellation? A whistleblower alerted Johnson & Johnson in 2006 to a risk estimate of one in 300—a number similar to FDA’s estimate—as well as a near-miss case, where a patient would likely have experienced an upstaging of her uterine cancer if she had undergone the procedure?
BV: With the morcellator, I understand from you (but have not otherwise confirmed) that FDA had raised the issue of upstaging with the manufacturers during the review process and discussed whether the labeling should address the issue. If so, because people were already concerned about upstaging, the company should probably have been looking for that. That’s an unusual situation, however.
My initial reaction to the “whistleblower” situation as you describe it, however, is that this was not a mandatorily reportable case. There was no actual case or event to report. The situation involved a potential future event, because in this case the morcellation was not performed. They did a standard surgical removal; found cancer cells, and realized that they had dodged a bullet.
FDA does not require reporting on “what might have been”—except where a device actually malfunctions without causing a reportable injury but if it similarly malfunctioned in another similar situation, it could have resulted in death or serious injury.
Think of an X-ray machine that unexpectedly exceeds its radiation emission control limit during a warm up, when no patient is exposed; had the same dose of radiation hit a patient, it would have killed him. In this case, the morcellator was not used and did not malfunction.
The underlying philosophy is that attention and limited resources should be focused on real problems, those actually seen. FDA, industry, and physicians should, of course, consider potential risks, but all would be overwhelmed if every conceivable risk was reported.
I know this philosophy is not satisfying to consumer safety advocates, but there are other systems that should address potential risks. A postmarket study of morcellator use in removal of fibroid tumors, looking for upstaging, might have been appropriate, for example. The Medical Device Reporting system, however, is to look at actual experiences.
Let me be very clear: I’m only saying that the “case study,” as you have described it, did not, in my view, trigger a legal obligation to report to FDA under the MDR regulations, because the device was not used and did not lead to an injury or malfunction, which are the essential predicates for an MDR reportable event.
In light of the pre-existing recognition of the risks of upstaging, the near-miss case should probably have led a manufacturer to reassess the potential risks, and perhaps led to warnings, to additional research, and to discussions with FDA. I’m not giving the company a full pass, just focusing on the MDR reporting question.
I’m going to jump ahead and circle back. Do you know if federal requirements for adverse outcomes reporting are different for drugs and devices?
BV: The regulations are different and they’re designed for different kinds of things, and the reporters are frequently different. There are differences in the basic structures of federal regulations, yes. They also differ in terms of what is a reportable event.
For drugs, there is no required reporting by anybody except the manufacturers. Hospital or as we call it, end user reporting, is only required for certain devices, partially because in the device arena, you have frequently a disconnect between the ordinary physician and the user facility—a lot of X-ray centers, MRI centers, things like that are independent of the doctor, and so the doctor would not necessarily know whether the machine had failed or things like that.
Another point is, the device regulations are looking for machine failures that would cause injuries if they occurred later on. In the drug arena, that’s not the way the formulation of language is used.
Individual practitioners are not held liable for reporting in the device or drug arena.
User facilities i.e. hospitals are required to report adverse outcomes resulting from medical devices—but why not drugs?
BV: I think it has to do with the special ability of the user facility to identify device-related events, at least as perceived at the time the regulations and laws were enacted. We are talking about specialized equipment like X-rays, MRI machines, CAT scans, radiation-beam therapeutics, etc.
If there’s a problem with the machine, the operators are more likely to know, and those are located in the user facilities, hospitals or MRI clinics—which would be required to report. The referring physician might not be likely even to detect a problem.
Now that doesn’t mean that hospitals don’t report drug-related adverse events. An Institute of Medicine report in the late 1990s discussed how many adverse events were caused by misprescribing, misdispensing and drug interactions, often in hospitals. As a result, many hospitals established risk committees to look into the utilization of drugs and adverse events. Now that hospitals have centralized identification of drug-related adverse events, they’re probably reporting more voluntarily than they did before.
Patient advocates are saying that individual practitioners should also be mandated to report. What do you think?
BV: It’s going to lead to a lot of litigation, as to when this or that doctor should have recognized the first case they had, and proving that the drug or device did this or that.
I’ve been involved in a number of litigations where they go after the company, and the plaintiffs always want to allege that the company should’ve known when the first case came in, that the drug was the cause of harm. And that, from a scientific standpoint, is rarely possible.
It is even more problematic for individual physicians. For every untoward outcome, she would have to determine, was it an accident, or part of the natural course of the disease, or drug-related? The default mode would be to report everything, regardless whether it was realistic. You don’t get in trouble for over-reporting, only failing to report.
Plus, consider the burden on FDA to police the medical community. Today, FDA has legal jurisdiction over the handling of food in most restaurants, yet relies on state and local food inspectors to inspect and monitor retail operations. FDA could not possibly find the resources to conduct routine inspections of physicians for possible failure to report an adverse event.
So you’re saying that it’s just too difficult because, generally speaking, individual physicians could be embroiled in a problem that they genuinely have no knowledge of?
BV: Remember, failure to report, on the federal level, is a crime. It’s not a civil thing like damages that your insurance companies pay for. It’s a crime. You want to encourage voluntary reporting, in which the doctor thinks about the case. If the only safeguard is to report everything bad, then public safety is not advanced by mandatory physician reporting.
A general philosophical standpoint is, the federal government, at least from FDA’s standpoint, does not try to regulate the practice of medicine or what doctors do. That’s left to the states.
Advocates also say that companies should be required to track the first wave of high-risk devices via a registry and report outcomes to FDA. Is this feasible?
BV: It is feasible, but it is very expensive. In the 1980s, the NIH established a patient registry for pacemakers; it died within 10 years, because the government could not afford to maintain it. In tracking patients, you have to get the doctors or hospital staff to fill out the paperwork (sometimes literally in the operating room), then collect the information and enter it into a database, and then follow the patients for an extended period of time. With the Unique Device Identifiers and electronic medical records, it’s going to become a lot easier to get accurate information on the implanted device and the patient, but somebody still has to transmit the information to the manufacturer (or registry operator), who must put the patient on the registry, and follow the patient through routine contacts with the patient or her treating physician.
This alone presents real issues of confidentiality. Patients might not want to be in the registry or otherwise refuse to cooperate. Years ago, an organization tried to set up a registry for breast implants; they met enormous pushback from women who did not want even their husbands to know they had a breast implant. If a researcher wants to test a hypothesis that involves getting non-anonymized patient information, HIPAA restrictions also kick in.
My bottom-line position is registries are generally not cost-effective. A company-oriented registry collects on its product, but not comparative data, because it’s not shared with other manufacturers. You can only have comparative data with a registry that covers all products in a certain category (e.g., pacemakers, hip implants) across the board. That’s a better way of doing it, and a number of independent registries covering all products in a given class (e.g., hip implants, heart rhythm devices) have been established by academic or professional groups. But if the manufacturers are going to participate in these arrangements, you have to resolve issues such as sharing the cost among companies, allowing access to the data, and determining whether data might be used for competitive (not health) purposes.
As you can tell, I am not a fan of registries today.
Is there a better way of doing it?
BV: The way to move forward, in my view, is a comprehensive medical device postmarket surveillance system that utilizes claims data and electronic health records. Just think about how medical reimbursement records could be used in the era of Big Data.
Let’s take morcellators as an example. Suppose you were to code (accurately and with high granularity) all patients who were treated with morcellators vs. other types of surgical interventions for the same medical conditions. It would be possible to probe the data to look for how many patients had treatments for uterine fibroid tumors and who subsequently were treated for uterine cancer in the next succeeding 12 or 24 months. You could then compare those treated with morcellators to those treated by other interventions. If—if—morcellation increased the risk for upstaging, it might appear very quickly from this type of query, with a minimum of cost and complications. It seems to me that if FDA and the manufacturer were concerned about the potential for this particular risk, instead of a registry or a small-sized postmarket study, they could agree on periodic probes into the medical reimbursement records to test the hypothesis. This approach would be much cheaper and faster than either a registry or a postmarket study.
Now and increasingly in the future, with electronic medical records and claims data, you don’t have to enter the patient into a registry of people. You can identify and study things no one has previously considered. With Big Data, you don’t need to collect the first 1,000 or 2,000 patients in a registry. That, to me, is the way of the future.
Let’s try and summarize. We’ve got two physicians-turned-patient-advocates, a controversial high-profile medical device, an FDA that acknowledges that this is a reporting of adverse outcomes issue, and a Congressional inquiry. What is the root cause of this problem, and what preemptive steps could have been taken to prevent the morcellator disaster?
BV: What the IOM report essentially said: FDA should focus more on risk at the time it’s looking at the products for preclearance. Move away from the simple 510(k) “substantial equivalence” evaluation, and concentrate more on the risks we know or associate with the device, and what are the risks we think could potentially be associated with the device.
And then, FDA should spell out the appropriate criteria for deciding whether to let that product and similar ones on the market. As I said, these criteria could be articulated in the context of groups of similar devices, rather than treating all Class II devices as a single group. In fairness, FDA does do this in practice, but the statutory standard of “substantial equivalence” applies to all Class II devices equally.
Once FDA has cleared the product, it and the manufacturer should both periodically revisit the product to determine whether there are safety concerns, either foreseen or not.
As the manufacturer makes modifications to the product, FDA should consider whether the modifications also respond to confirmed safety issues. Much of the burden for answering these questions will lie with the manufacturer, of course.
Basically, the IOM Committee wanted to introduce the concept of risk-management throughout the lifecycle of the device as a regulatory requirement. You’re not going to prevent all bad things from happening. What you hope to do is to reduce the number of casualties and the duration before you catch the problem at fix it.
That’s your goal. Detect it early and intervene to prevent further harm.
So that’s the preemptive part. Do you think the federal mandate for self-reporting right now is the best that it can be?
BV: Yes, unfortunately. I think we’ve had a lot of experience with self-reporting in the drug arena, and to some extent in the device arena. It’s just very difficult to collect a lot of good data.
When you look back at the last 40 years, you can see that we’ve been unlucky in that we missed some problems for years or even decades, that it took a long time to recognize that this drug or that device was causing a health problem, and then to figure out why and how it was causing that problem.
I don’t see any major improvements in the voluntary self-reporting system that will vastly improve our ability to detect and fix problems, though I hope that some of the initiatives underway at FDA and in the medical community will make a difference.
Voluntary reporting remains valuable for providing insight. The guy who says “I think this may be a problem” often has thought long and hard about it, and has generated a hypothesis.
But to test such a hypothesis, we need to move from the current system to Big Data, where you take a huge amount of claims data and link it up with particular devices, and then look at the health claims filed by patients in the months of years later to see what happens after a particular device put in or used on them, in comparison to other similar devices or to other forms of treatment not involving a device. Instead of going to the spontaneous reporting database FDA can go to claims data and say, “Let’s run this through the computer and get results.”
If there is upstaging of cancer, and if this is more commonly associated with use of a particular drug or device over others, you should be able to see these trends more rapidly because you have a huge amount of data. From 2015 to 2025, I would argue that Big Data is the way we’re going to head.
Registries will become a thing of the past, and we won’t have to rely on voluntary reporting terribly much.