FDA’s passive reliance on self-reporting by hospitals and device manufacturers allowed harm caused by power morcellators to go unnoticed for over two decades—likely contributing to injury and deaths of hundreds of women, according to the U.S. Government Accountability Office said.
The power morcellator presents a unique case study in patient harm, said Marcia Crosse, director of the health care team at GAO.
The power morcellators worked as they were intended to work: they did not fail at their intended use of morcellating large tumors. As a result, some hospitals interpreted the language of the statutory requirement to report adverse events as not applicable to the dissemination of malignant tissue. FDA has debunked this contention, saying that such cases should, in fact, have been reported.
Physicians need to start thinking of adverse events in the context of a device that functions as intended, GAO’s Crosse said in a conversation with The Cancer Letter, in which she elaborated on the findings of a GAO investigation.
GAO recently released a 49-page report that sums up the three-year controversy over the once-common minimally invasive surgical procedure, now believed to have disseminated unsuspected uterine cancer in about one in 350 women.
“If the device had broken in the middle of a surgery, if the device had left a piece behind, the tip had fallen off or something like that, I think that the medical establishment is used to thinking of those kinds of device-related events as adverse events to be reported,” Crosse said. “They may not be accustomed to thinking—and they need to think more broadly—that the device can work as it was intended to work and still cause harm.
“I think that was not, perhaps, being recognized sufficiently. There are folks in large medical centers charged with responsibility for this reporting, and so I’m hoping this was a wake-up call.”
The conversation with Crosse appears here.
The GAO report evaluated FDA’s clearance process for power morcellators as well as the agency’s handling of the hazards these devices posed to public health. The watchdog agency prepared the report in response to a request by 12 members of Congress to investigate why “hundreds, if not thousands, of women in America are dead.” The House members requested the investigation in August 2015.
“Certainly, the hundreds seem reasonable, I cannot say thousands,” Crosse said to The Cancer Letter. “While we’re aware of 285 adverse event reports that were filed with the FDA [since December 2013], we are not making any independent estimate of the number of women harmed.”
The hazards of power morcellation made national headlines after one patient, Amy Reed, an anesthesiologist and mother of six, underwent the procedure at Brigham & Women’s Hospital in October 2013. The device’s spinning blades had spread her undetected leiomyosarcoma, an aggressive uterine cancer, during the surgery. Reed is being treated for stage IV disease.
There are no authoritative estimates for the number of women who have died as a result of power morcellation. Over 80 percent of black women and nearly 70 percent of white women develop fibroids in their lifetime. According to studies, between 50,000 to 100,000 women a year in the U.S. underwent the procedure before FDA severely limited the use of power morcellators for hysterectomies and myomectomies in November 2014.
Summary of Adverse Event Reporting Requirements for Medical Device Importers, Manufacturers, and User Facilities | ||
What to report | To whom | When |
Importers | ||
Deaths and serious injuriesa | FDA and the manufacturer | Within 30 calendar days of becoming aware of an event |
Malfunctionsb | Manufacturer | Within 30 calendar days of becoming aware of an event |
Manufacturers | ||
Deaths, serious injuries, and malfunctions | FDA | Within 30 calendar days of becoming aware of an event (or within 5 work days upon FDA’s request) |
Deaths, serious injuries, and malfunctions requiring remedial action | FDA | Within 5 work days of becoming aware of an event |
Supplemental reports to provide new, changed, or corrected information for a previously submitted report | FDA | Within 30 calendar days of receipt of the information |
User facilityc | ||
Death | FDA and manufacturer, if manufacturer is known | Within 10 work days |
Serious injury | Manufacturer, or FDA if manufacturer unknown | Within 10 work days |
Annual summary of death and serious injuryd | FDA | January 1 for the preceding year |
aSerious injuries are injuries or illnesses that are life-threatening, result in permanent impairment of a body function or permanent damage to a body structure, or that necessitate medical or surgical intervention to preclude permanent impairment of a body function or damage to a body structure. bMalfunctions are defined as the failure of a device to meet its performance specifications or otherwise perform as intended. cA user facility is a hospital, ambulatory surgical facility, nursing home, or outpatient treatment or diagnostic facility that is not a physician’s office. dUser facilities are required to file annual reports that summarize their adverse event reports. | ||
Source: FDA | GAO-17-231
GAO notes that FDA cleared 25 versions or submissions for power morcellators between 1991 and 2014 to be marketed in the U.S. Critics of power morcellation say this is evidence that hundreds or thousands of patients have been harmed or have died from upstaged metastatic disease in that period.
Despite evidence that device manufacturers and physicians at prominent hospitals knew that patients were being harmed by power morcellation, nobody reported these adverse events to FDA—an action required of user facilities and manufacturers by federal law—until Reed reported her case in December 2013 (The Cancer Letter, Nov. 20, 2015).
“I do think it was a failure of the adverse event reporting system, yes,” Crosse said. “I think it’s a failure because reports were not being filed.”
In an investigative series, “How Medical Devices Do Harm,” The Cancer Letter broke stories in 2014 and 2015 examining FDA’s system for reporting adverse events, and found that physicians at Brigham & Women’s Hospital and Johnson & Johnson officials had direct knowledge of patient death and harm years before Reed’s highly publicized account.
In December 2015, FDA initiated inspections at 17 hospitals to review their compliance with medical device reporting requirements. The vast majority of those hospitals did not file timely reports of injuries and deaths caused by medical devices. However, the agency decided against taking punitive action. (The Cancer Letter, Dec. 16, 2016).
“These inspections included five hospitals that, according to FDA officials, were chosen because there were reports of adverse events at these facilities related to the spread of uterine cancer from the use of power morcellators,” the GAO report states.
The GAO investigation found that FDA’s system for reporting adverse events has the following limitations:
Incomplete or erroneous reporting. Adverse event reports can include incomplete reporting, where key data are not reported, or erroneous reporting, where the information provided is not accurate.
Reports that are not timely. Adverse event reporting does not always reflect real time reporting, as some reports document events that occurred years earlier.
Underreporting. Adverse events may not always be reported.
Members of Congress are planning on taking the issue to the White House. Rep. Brian Fitzpatrick (R-PA), who succeeded his brother Mike Fitzpatrick in the recent election, is leading the charge, his office confirmed.
Fitzpatrick plans to reintroduce the Medical Device Guardians Act, which would require individual physicians and practitioners—instead of only at the hospital administration level—to report adverse events.
“There are serious gaps in the FDA’s device reporting system and immediate Congressional action is needed to reform the process and save lives,” Fitzpatrick and Rep. Louise Slaughter (D-NY) said in a statement. “Armed with this information, we will move forward to find bipartisan legislative solutions to address these shortcomings and ensure a system is in place that provides real, accurate information to patients, professionals and regulators.”
FDA knew of the risk of dissemination since 1991
The GAO report notes that FDA officials were aware of the potential for spreading tissue—cancerous or noncancerous—during procedures that involved the use of power morcellators since the agency cleared the first device in 1991.
“We found that this awareness was reflected in the labeling for 12 of the 25 devices cleared by FDA,” the report states. “Agency officials, however, noted that [prior to December 2013], there was no consensus within the clinical community regarding the risk of this occurring, particularly for cancerous tissue.
“FDA officials stated that prior to December 2013, the general understanding was that the risk of a woman undergoing treatment for fibroids having unsuspected cancer—specifically, a difficult to diagnose cancer called uterine sarcoma—was low. FDA officials were not aware of any definitive scientific publications regarding the actual risk of cancer in uterine fibroids.”
After Reed and her husband Hooman Noorchashm, a cardiothoracic surgeon, launched an aggressive campaign to stop the use of the procedure, FDA conducted a review of published and unpublished literature, including patients operated on from 1980 to 2011, to determine the risk of spreading unsuspected cancerous tissue.
“This report makes it an indisputable fact that a professional and corporate failure in gynecology left the severe oncological hazard of power morcellators unreported to the FDA,” Noorchashm said to The Cancer Letter. “The GAO clearly documents that federal reporting requirements directed at hospitals and manufacturers were violated, leading to a sustained level of harm for two decades.
“But the report also highlights that, with adequate expert reporting, FDA can and does act to protect the public. It is a fact confirmed by the GAO, that our [adverse event] report in December 2013 triggered the FDA into action. So adequate expert reporting could very clearly serve to curtail dangers in the medical device space.”
According to Columbia University researchers, the use of morcellators has dropped by nearly 80 percent after FDA’s guidance document in November 2014 contraindicated the device for hysterectomies or fibroid removal in the vast majority of women getting these procedures.
Gynecology is now in a “post-morcellator era,” Amanda Nickles Fader, associate professor and director of the Kelly Gynecologic Oncology Service at Johns Hopkins, wrote in an email to Noorchashm, who routinely makes his correspondence dealing with the scientific and policy issues on morcellation available to reporters and regulators.
“The medical community must find a solution to the morcellation issue so that more women do not suffer in the same way Amy [Reed] has, while aiming to strike a balance and maximize MIS in the hundreds of thousands of women who will benefit from it,” Fader wrote in the email to Noorchashm. “We know that a large shift away from MIS will lead to great morbidity, and even mortality, in women.
“But we also recognize that we are in a post-morcellator era, and I (and many other investigators) are committed to developing better and safer technology so that we can avoid dissemination of occult malignancies.”
Fader did not respond to this reporter’s request to discuss her comments further.
“Dr. Fader’s position that we are currently in a ‘post-morcellator era’ is an indication that many leading gynecologists see the serious problem with this practice, and are moving courageously in the direction of taking ‘universal oncological precautions’ to protect all women from cancer upstaging,” Reed and Noorchashm said. “But unfortunately, so long as this device stays on the market, more women will be harmed by gynecologists who do not share Dr. Fader’s sentiment.”
Crosse cites failure to produce risk estimates of cancer
FDA concluded in April 2014—two decades after power morcellators started to be used for gynecologic laparoscopic procedures—that approximately one in 350 women undergoing hysterectomy or myomectomy for the treatment of fibroids is found to have an unsuspected uterine sarcoma.
“I think the problem here was a failure to have an accurate estimate of the frequency with which there is an undiagnosed leiomyosarcoma in what is believed to be a uterine fibroid, a benign fibroid,” Crosse said.
Some FDA insiders say that input from cancer experts within the agency into the review process for medical devices might have prompted FDA to prioritize studying the risk sooner, potentially saving lives. While the agency’s centers do collaborate, products are largely reviewed and regulated separately according to category: drugs, devices, and biologics.
An intercenter institute, the FDA Oncology Center of Excellence was established in June 2016 as a part of then-Vice President Joe Biden’s National Cancer Moonshot Initiative to streamline and consolidate regulation of cancer-related products. Richard Pazdur, director of the FDA Office of Hematology and Oncology Products, was recently named director of the OCE (The Cancer Letter, Jan. 20).
“It is a significant analytical error on the part of the GAO to state that the problem was a ‘failure to have an accurate estimate’ of occult sarcoma,” Noorchashm said. “How would knowing the ‘one-in-however many’ incidence address the fact that no one reported this danger to FDA?
Certainly, the hundreds seem reasonable, I cannot say thousands. I do think it was a failure of the adverse event reporting system, yes. I think it’s a failure because reports were not being filed. There are folks in large medical centers charged with responsibility for this reporting, and so I’m hoping this was a wake-up call.
“The only relevant failures that led to the sustained level of harm caused by power morcellators for over 20 years were: 1) a professional and corporate non-compliance with federal public health law by failing to report specific cases of cancer upstaging to FDA, of which there were many, and 2) the failure of leading gynecologists to think critically and ethically about this practice and its deadly but avoidable hazard to their specific patients.”
FDA officials said they agreed with GAO’s findings, and that FDA has noted the limitations of the existing system for reporting adverse events.
“The FDA has reviewed the GAO report on laparoscopic power morcellators and agrees with its findings,” agency officials said in a statement to The Cancer Letter. “To reduce the risk of spreading unsuspected cancer, the FDA continues to warn against the use of laparoscopic power morcellation for the vast majority of women undergoing the removal of the uterus or removal of uterine fibroids.
“A boxed warning for laparoscopic power morcellators states that uterine tissue may contain unsuspected cancer and that patients should be informed of the risk of spreading cancer from the use of these devices during fibroid surgery. The FDA continues to review information on laparoscopic power morcellation, including the latest data and evolving scientific literature, and will communicate publically if the agency’s recommendations change.
“In addition, the FDA has noted the shortcomings of the current passive postmarket surveillance system and has been taking steps to establish a better system to evaluate device performance in clinical practice. For example, the agency has awarded a grant to the Medical Device Innovation Consortium to establish a coordinating center for the National Evaluation System for health Technology (NEST). NEST is intended to improve the quality of real-world evidence that health care providers and patients can use to make better informed treatment decisions.”
GAO did not include recommendations for further action, because investigators believe FDA adequately addressed the crisis.
“We felt that FDA had taken and was taking appropriate steps. They had put out information about this,” Crosse said. “They had requested labeling changes from manufacturers and manufacturers had made those labeling changes. And they had undertaken a review to develop a better estimate of the underlying risk. They had taken the kinds of steps that we potentially could have recommended, had they not already been done.”
FDA enforcement of reporting requirements
Will FDA’s new surveillance system—which will require at least five years of development before it is fully functional—improve the agency’s ability to track and quickly address significant adverse events?
“I think it’s too soon to know how much more effective it will be,” Crosse said.
In the meantime, Rep. Fitzpatrick’s Medical Device Guardians Act, which requires individual physicians to report adverse events, would be effective for catching signals of harm, Noorchashm said.
“In an ideal world, one would design robust active surveillance frameworks for devices to detect dangers,” Noorchashm said. “But such a surveillance system will require several years of labor and cost intensive work for FDA to design and implement—not to mention the need for even more regulations to impose such as system. And even then outcomes surveillance systems would require active data mining to identify significant problems.
“So it stands to reason that enhancing reporting requirements, directed specifically at expert practitioners (as is missing from current regulation), would be a powerful stop-gap measure to provide FDA with the high fidelity ‘intel’ it needs from the ground to contain and curtail hazards to public health in the medical device arena. Such a system would be relatively cost-neutral, as a reporting framework and database already exists at the FDA and was the pathway through which the power morcellator disaster came to light.”
It’s unclear whether requiring individual practitioners to report adverse events would have prevented harm from power morcellators, Crosse said. She declined to comment on the Medical Device Guardians Act.
FDA’s investigation and the warning letters that they issued showed that there was more room for enforcement of those [reporting] requirements, because, clearly, there was a failure to comply at many institutions.
“That’s speculation, I don’t know. I think if FDA had been aware of it sooner, it could have taken action sooner,” Crosse said. “I don’t know if the entire problem could have been avoided by that.”
FDA does not have the resources to conduct regular inspections to ensure hospitals are compliant with reporting requirements, Crosse said.
“[The agency has] been reliant upon the good faith and execution of those requirements by the medical establishment,” she said. “I think FDA’s investigation and the warning letters that they issued showed that there was more room for enforcement of those requirements, because, clearly, there was a failure to comply at many institutions.”
Following the December 2015 inspection of 17 institutions, FDA imposed no penalties against the hospitals that failed to comply with the reporting requirements, because “these hospitals indicated their willingness to work with us and address the violations,” officials said at the time (The Cancer Letter, Dec. 16, 2016).
For reporting requirements to be effective, adequate law enforcement by FDA is an “absolute necessity,” Noorchashm said.
“If hospitals and manufacturers are found to be in definitive violation of federal reporting requirements, as is the case here, a letter of warning from FDA is fully insufficient, especially when unsuspecting patients have been harmed or died as a result,” Noorchashm said. “The FDA’s law enforcement unit ought to impose significant fines or recommend prosecutions by the U.S. attorney general or inspector general of HHS for wrongful deaths in such cases.”
Patients with leiomyosarcoma have a 5-year relative survival rate of 63 percent when diagnosed at stage I, according to the American Cancer Society. At stage IV, the survival rate drops to 14 percent.
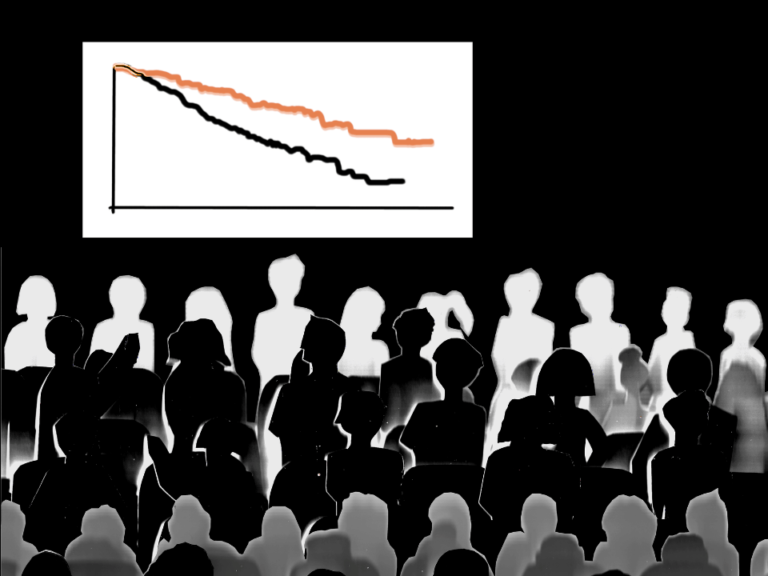
A retrospective cohort study of 58 patients, published in the journal Cancer, found that patients who underwent morcellation were almost four times more likely to have a recurrence of malignant disease. The median recurrence-free survival for these patients was significantly shorter compared to those who underwent total abdominal hysterectomies (10.8 vs. 39.6 months).
Patients who underwent morcellation were twice as likely to die from metastatic disease—the median overall survival was 48 months—than patients who had total abdominal hysterectomies.
Since Reed’s report in December 2013, a number of patients who underwent power morcellation and their family members shared their stories with the press. Following is a partial list of women who have died in recent years from metastatic uterine cancer upstaged by the procedure:
Martha Ariri, Riverside, Calif. Chicago Tribune
Nancy Curtis, Missoula, Mont. KPAX.com
Bonnie Davis, Pittsburgh Pittsburgh Post-Gazette
Linda Interlichia, Brighton, N.Y. The Wall Street Journal
Elizabeth Jacobson,Sacramento, Calif. The Washington Post
Erica Kaitz, Boston The Cancer Letter
Barbara Leary, Greece, N.Y. Democrat & Chronicle
Brenda Leuzzi, Perinton, N.Y. Democrat & Chronicle
Viviana Ruscitto, Nyack, N.Y. The Record/NorthJersey.com
Danusia Taber, Newbury Park, Calif. CBS Los Angeles
ACOG: All medical procedures carry risk
Risk can never be completely eliminated, and power morcellation remains an important option for women, said Hal Lawrence, executive vice president and CEO of the American College of Obstetricians and Gynecologists.
“ACOG applauds the U.S. Government Accountability Office on its recent report regarding the use of power morcellators in minimally invasive gynecologic surgery,” Lawrence said in a statement. “The report presents a thorough investigation of the issue, and ACOG shares in the federal government’s commitment to provide the best evidence to patients regarding the safety of using power morcellators to treat uterine fibroids.
“While the device has faced scrutiny in recent years, as the leading organization representing health care providers for women, ACOG maintains that power morcellation is an important option for women undergoing surgery for uterine fibroids. Morcellation is a minimally invasive technique that spares women from increased morbidity and mortality associated with abdominal surgery.
“Of course, when considering any procedure, ACOG recommends that ob-gyns conduct a thorough patient evaluation, engage in shared decision making, and follow the informed consent process with each patient. In the case of morcellation, this also includes reviewing appropriate measures to evaluate risk factors for potential malignant uterine sarcoma before moving forward with this treatment option.
“All medical procedures carry risk and that risk can never be completely eliminated. Moving forward, ACOG welcomes the collection of meaningful data that will help provide for the safe and effective use of power morcellation.”
The GAO report is thorough and reflective of the history surrounding power morcellators, according to the American Association of Gynecologic Laparoscopists.
“The AAGL has reviewed with interest the Bipartisan Group Release GAO Report on Medical Device Safety,” AAGL officials said in a statement. “The GAO has evaluated in detail the process by which the FDA reviewed 510(k) submissions for power morcellators, the FDA’s understanding of any concerns surrounding the use of power morcellators, and the professional standards and guidance for physicians using these devices.
“We believe that the FDA fulfilled its responsibility in reviewing the devices, investigating reports of adverse events, providing education surrounding its findings, incorporating information into required product labeling, and following up with organizations and hospitals to ascertain uptake of their recommendations.
“The AAGL remains supportive of products and techniques that improve the outcomes of women undergoing minimally invasive surgery for gynecologic care and appreciates the FDA’s commitment to ensure the safety of approved devices.”
Timeline of Key Events Related to Laparoscopic Power Morcellators | |
Date | Key event |
March 1988 | The Food and Drug Administration (FDA) clears the PacesetterTM 3500 Arthroscopic Surgical System—a predicate device for the first power morcellator—for the U.S. market. |
June 1991 | FDA clears the Cook Tissue Morcellator—the first power morcellator—for the U.S. market. |
May 1995 | FDA clears the KSEA Steiner Electromechanic Morcellator—the first power morcellator with indications for use for gynecologic laparoscopic procedu res—for the U.S. market. The indications for use specifically identified the removal or morcellation of uterine fibroids. |
February 2000 | FDA clears the Ethicon Gynecare Laparoscopic Morcellator—the first power morcellator with indications for use that identified hysterectomies (among other procedures)—for the U.S. market. |
November 2013 | FDA receives the first notification of an event where the use of a power morcellator during surgery to treat uterine fibroids may have spread an unsuspected uterine cancer. |
December 2013 | The Wall Street Journal publishes an article on the same event. FDA receives the first adverse event reports of the spread of unsuspected uterine cancer following the use of a power morcellator. In response, the agency convenes a signal review team to coordinate and lead FDA’s evaluation and response to the potential power morcellator safety issue. |
April 2014 | FDA publishes the results of a review of scientific literature published since 1980, and finds that the risk of having an unsuspected and difficult to diagnose type of cancer, uterine sarcoma, is about 1 in 350 for women undergoing the surgical procedures of hysterectomy or myomectomy to treat uterine fibroids. FDA also estimated that the risk for having a specific type of sarcoma called leiomyosarcoma was about 1 in 500 among such women. FDA issues a safety communication that (1) reports the higher rate of unsuspected uterine cancer in women who undergo treatment for uterine fibroids (about 1 in 350), and (2) discourages the use of power morcellators in surgical procedures (hysterectomy or myomectomy) to treat uterine fibroids. FDA also sends letters to power morcellator manufacturers strongly recommending the review of product labeling and coordination with the agency to ensure that such labeling addresses the estimated risk. |
July 2014 | FDA convenes a meeting of the Obstetrics and Gynecology Devices Panel of FDA’s Medical Devices Advisory Committee to solicit stakeholder input and available data related to the potential power morcellator safety issue. One manufacturer of power morcellators initiates a voluntary withdrawal of its power morcellators from the U.S. market. |
November 2014 | FDA issues an updated safety communication and an “immediately in effect” guidance recommending manufacturers include a boxed warning and additional contraindications in their product labeling. FDA’s guidance states that manufacturers should implement these labeling recommendations and that within 120 days, a manufacturer with an existing 510(k) clearance should (1) add the contraindications and boxed warning to their labeling; (2) submit revised labeling to FDA; and (3) provide updated labeling to purchasers for power morcellators that have already been distributed. The safety communication also states that FDA considers the spread of an unsuspected cancer following the use of a power morcellator to treat uterine fibroids as a serious injury reportable under adverse event reporting regulations. |
December 2015 | FDA initiates inspections at selected hospitals to review their compliance with medical device reporting requirements. These inspections included five hospitals that, according to FDA, were chosen because there were reports of adverse events at these facilities related to the spread of uterine cancer from the use of power morcellators. Enrollment begins in the COMPARE-UF registry phase, which is expected to enroll about 10,000 women and evaluate the effects of treatments for uterine fibroids. |
April 2016 | FDA permits the marketing of a new type of device, a tissue containment system that could be used with certain power morcellators during morcellation of noncancerous uterine tissue for certain patients. FDA required the manufacturer of the new tissue containment system to warn patients and health care providers that the system has not been clinically proven to reduce the risk of spreading an unsuspected uterine cancer. |
Source: GAO | GAO-17-231