Researchers are coming to the conclusion that cancer patients are the best authority on how therapies affect them.
Over the past few years, about 20 NCI trials and over 125 industry studies have incorporated direct reports of symptomatic adverse events by cancer patients through an NCI program.
Last year, building on Vice President Joe Biden’s 2016 Cancer Moonshot, NCI issued a Request for Applications to study application of patient-reported outcomes. The institute said it intends to commit $3.25 million in FY2018 to fund three to five awards.
Here is how such data can be used: (1) investigators predict a therapy’s side effects, (2) they list these side effects in an electronic questionnaire, (3) patients report on their experience, and (4) investigators refine their understanding of the way the drug is tolerated and, possibly, adjust the regimen.
Some studies have shown that reporting of symptomatic adverse events by patients is more reliable than such assessments by physicians. Two oncologists seeing the same patient minutes apart from each other tend to disagree quite frequently about the level and severity of symptoms.
Through patient-reported outcomes, clinicians would be able to improve the measurements of risk and benefits, said Ethan Basch, director of the Cancer Outcomes Research Program and professor of medicine and public health at the UNC Lineberger Comprehensive Cancer Center.
“There was previously a bias among some investigators that ‘I know better than my patients,’ or that we can’t really trust patients to be reliable, or that patients will be too ill to report their own toxicties, but empiric data show that none of that it true,” Basch said to The Cancer Letter. “What we find is that, in fact, when clinicians report patient symptoms, that’s where the real subjectivity comes in.
“Without patient-reported information about these experiences, we have an incomplete understanding of the characteristics of drug products. And that simple messaging, I hope, will really push the industry to bring their resources forward around the methods for doing this.”
A conversation with Basch appears here.
Recording patient-reported outcomes electronically in real time and allowing clinicians to review longitudinal reports can improve patients’ quality of life and lengthen survival, Basch wrote in an article published January 2017 in the New England Journal of Medicine.
More than a decade ago, many in the oncology field scoffed at efforts by Basch and his team to systematize these data by using an existing grading scale for adverse events.
“I faced a lot of skepticism from my colleagues, who, first of all, said, ‘We understand our patients well, so we don’t need them to provide their own information. We know what’s going on with them, we talk to them all the time,’” Basch said. “And the second [strand of] skepticism that I faced was that colleagues felt that it just wasn’t feasible to collect this information.
“We demonstrated conclusively that that just wasn’t the case. We found the vast majority of patients were willing and able to self-report even when they were extremely ill and even close to death or hospice.”
Now, at NCI and in the pharmaceutical industry, cancer research is moving beyond basic clinical metrics for drug safety, to including patient self-report measures—enabling investigators to assess patient outcomes over time.
These self-reports allow for real-time capture of symptomatic adverse events experienced by patients undergoing cancer treatment, giving researchers insight into the patient experience outside the clinic and providing additional data on the frequency and severity of the toxic effects of investigational drugs.
Enter NCI’s patient self-reporting platform, PRO-CTCAE, also known as the Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events.
The program was built as a companion to the institute’s existing CTCAE library, which is a standardized set of criteria used to classify adverse events from drugs used to treat cancer. Before the PRO companion was developed, clinical investigators relied on the CTCAE to grade the severity of each adverse event, based on laboratory findings, clinical observations and other sources of data.
The PRO-CTCAE item library has been publicly available since April 2016, eight years after the start of its development.
Conceptually, the CTCAE bundles the frequency and severity of adverse events, whereas the PRO component serves to distinguish the frequency, severity, and interference associated with each of those toxicities.
The CTCAE provides a comprehensive list of adverse events—about 800, each graded on a five-point scale—its use generates safety data on patients at limited treatment cycles. On its own, the CTCAE doesn’t allow investigators to assess the onset, resolution, or trajectory of toxicities or symptomatic adverse events resulting from therapy over time.
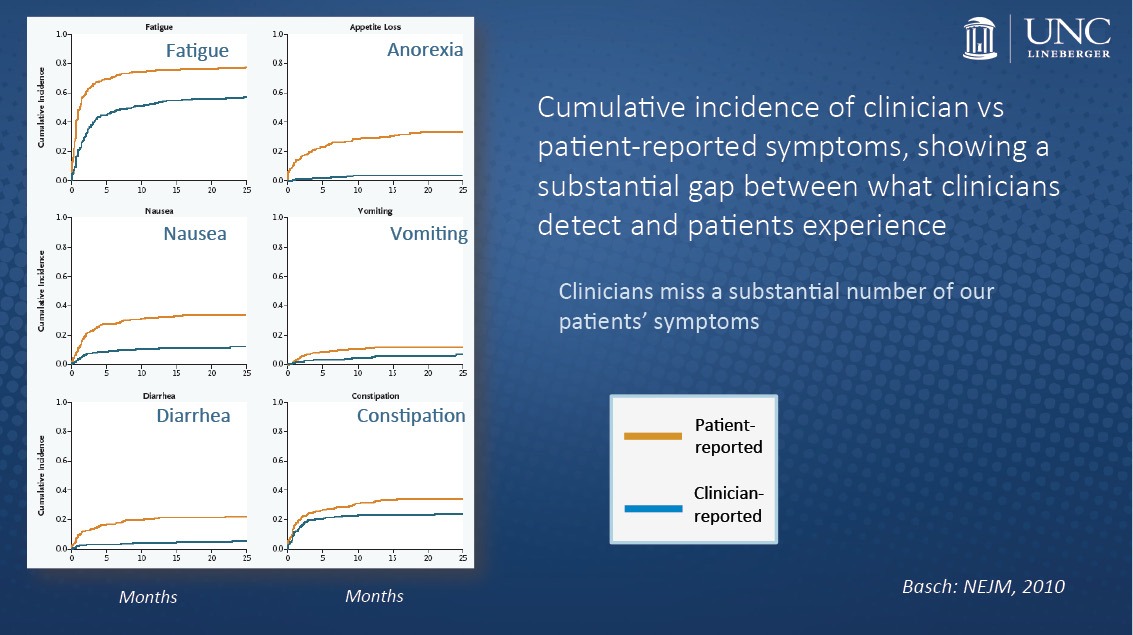
“The routine collection of toxicities by clinicians didn’t fully capture the patient experience and often missed some of the problems that were of most importance to patients,” Barnett Kramer, director of NCI’s Division of Cancer Prevention, said to The Cancer Letter. “These occurred between study visits, or were chronic, and therefore not picked up in any specific clinical visit. So, it was known that on questioning, patients would very clearly state that they had either more frequent, more chronic, or more severe symptoms than were captured in the medical record.”
Self-reports are used not only to examine toxicity, which would enable investigators to address the need for action or management of particular adverse events.
The Moonshot RFA will focus on using PRO-CTCAE with CTCAE data and other clinical trial data to determine tolerability.
“The initiative started sometime around 2014 and the [Moonshot] RFA shows that it’s still an evolving process to refine it and make sure that it is fully implemented,” Kramer said.
The application period for the RFA closed on Jan. 17. At this time, the RFA was a one-time solicitation, but investigators are also free to submit investigator-initiated awards, NCI officials said.
“There’s a collaboration that has gone on with FDA, as far as I’m aware, from the beginning of the development of PRO-CTCAE, because the information clearly could be of real use to the FDA, not just the NCI research community and to clinical practitioners, but ultimately could be in the mix of how the FDA looks at drugs,” Kramer said.
“But the main thrust of the funding opportunity announcement was to enhance our understanding in the cancer institute of how it’s used, who uses it, the utility, and simple things like, how can the different domains of the PRO-CTCAE be combined and analyzed, and whether or not the same questions can be modified for children.”
Safety vs. tolerability
The primary goal for PRO-CTCAE is to improve researchers’ understanding of patient tolerability—how well patients can tolerate a treatment regimen or investigational agent.
“I think that safety and tolerability overlap, but there’s not a one-to-one correspondence,” Kramer said. “Very frequently, the clinically-obtained side effects are used directly in protocols to adjust doses and especially in early phase studies, phase one studies, to find the maximal tolerable dose for the patient.
“But, the goal of the PRO-CTCAE is to go beyond that and get at the full patient experience and their tolerability over time, and so not every patient can tolerate the maximally tolerated dose or one dose below that. Compliance may be much, much worse in the real world than would be suggested by simply delivering the maximally tolerated or safe dose.
“I think they’re complementary. The PRO-CTCAE provides information not obtained through CTCAE and vice versa, so I don’t see one eliminating the need for the other.”
These complementary measures will become increasingly important with the development and testing of immunotherapies and molecularly targeted agents.
“Our vision for PRO-CTCAE in NCI clinical trials is to capture patients’ perception of symptomatic adverse events in a manner that’s complementary to clinician-graded adverse events, the primary reason of which is really to improve our identification of tolerability for cancer treatment regimens and cancer prevention and control interventions,” Lori Minasian, deputy director of NCI’s Division of Cancer Prevention, said at a recent joint meeting of the NCI Board of Scientific Advisors and the National Cancer Advisory Board.
“Patients are receiving drugs on a chronic basis, with some of these toxicities developing into chronic toxicities and things that impair adherence over the long run,” Minasian said. “PRO-CTCAE data currently is analyzed like other PRO data—analyzed in aggregate, out of trial-level, to provide information about the trajectory over time, but really does not result in any dose modification for an individual patient at this time.
“There’s a huge opportunity here to use this brand-new tool to provide a signal for tolerability that will give us more information on how to use this and how to think about delivering therapy to patients.”
NCI’s ongoing work is focused on interpretability and utility, said Sandra Mitchell, a research scientist at the Outcomes Research Branch, Healthcare Delivery Research Program at NCI’s Division of Cancer Control and Population Sciences.
“Measuring safety and tolerability in cancer clinical trials is really fundamental to the kinds of conclusions we wish to draw about the effectiveness of cancer therapies,” Mitchell said at the meeting. “This is really the motivation for us to support the development of the Patient-Reported Outcomes version of the CTCAE, and it’s designed to allow for real-time ascertainment using PROs to improve the precision and reproducibility of our capture of symptomatic adverse events.
“We’ve built this tool, but we really need to understand better how best to interpret it and what is its clinical utility. This is still evolving. We have broad implementation in a number of early-phase studies and randomized studies, both NCI-sponsored trials but also, we’ve had very broad adoption of this through pharma, and this work has definitely been encouraged and supported through our interactions with the FDA.”
Patient-reported outcomes may one day become a factor in drug approval. Alongside other data, these reports may also be used as a basis for dose modifications or treatment discontinuation, said Paul Jacobsen, associate director of the Healthcare Delivery Research Program at the DCCPS.
“There is good reason to believe that the validity of symptom toxicity reporting could be strengthened by the inclusion of patient self-report measures,” Jacobsen said at the meeting.
“We’ve drawn from 78 symptomatic adverse events listed in CTCAE to develop an item library of questions and items, and it’s possible to create customized surveys so that not every patient sees every item, but only those items that are related to the potential toxicities from the treatments they might receive.
“Work on PRO-CTCAE has progressed to the point that has been publicly available since April 2016, along with related materials.”
Real-world implementation
Systematic collection of patient-reported outcomes outside the clinical trial setting is technically challenging, because electronic health record systems are evolving slowly, Basch said.
“There’s quite a bit of interest not just in oncology, but beyond to systematically collect patient reported outcomes during clinical care,” Basch said. “Electronic health record vendors have been slow to develop PRO interfaces well, which has stalled the widesp
I think that safety and tolerability overlap, but there’s not a one-toone correspondence. Very frequently, the clinically-obtained side effects are used directly in protocols to adjust doses and especially in early phase studies, phase one studies, to find the maximal tolerable dose for the patient.
Barnett Kramer
read collection of this information. There has been a lot of pressure on the big EHR vendors to improve their PRO functionality.”
Real-world data is playing a rapidly expanding role in cancer research—by tracking drug use and effectiveness in the real world, in real-time, researchers and drug manufacturers are able to respond quickly to adverse events and also aggregate patient data to inform regulatory decisions.
“Well, there are multiple reasons why it’s important to collect this information in the real world,” Basch said. “One reason is to manage patient symptoms; the second reason is to collect population level data to understand problems that people have long-term that we simply can’t collect in very small, brief, clinical trials.
“And then the third is to do comparative effectiveness research, where we compare therapies to each other based on population-level data. And currently, we don’t have a living repository of patient-reported outcomes to do effectiveness research—this could be a future area for the NCI’s SEER registries to develop.”
A best practices white paper for the PRO-CTCAE is needed, and ideally would be developed in partnership with clinical investigators, PRO experts, as well as NCI and FDA, Basch said.
“It probably should come through a multidisciplinary meeting that is sponsored by the NCI that brings together outside investigators, regulatory authorities, patients, and other stakeholders in order to lay out how this tool should be used,” Basch said. “I laid out for you how I feel it should be used in a trial, but that needs to be codified with the imprimatur of multiple stakeholders.”