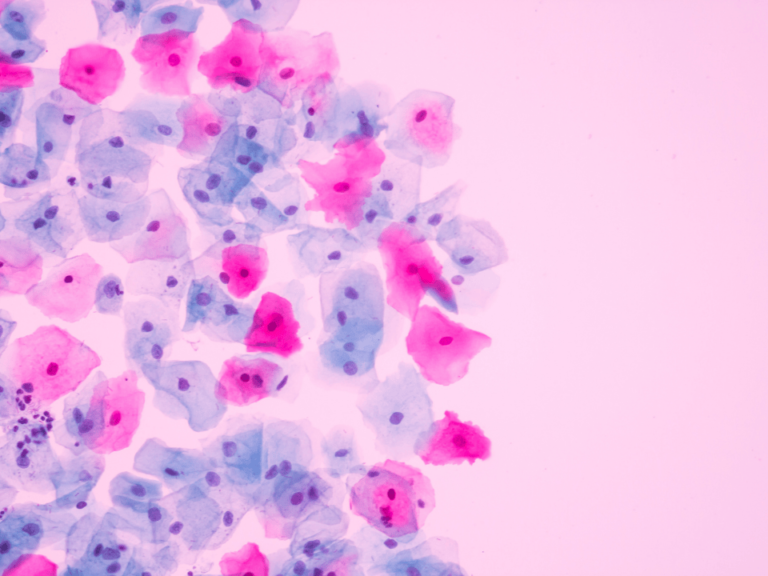
The Government Accountability Office Feb. 8 released an analysis of FDA’s failure to detect the health hazards of power morcellation, a once widely used procedure that has been shown to upstage uterine cancers.
The upstaging has been estimated to occur in about one in 350 women.
GAO notes that between 1991 and 2014 FDA cleared 25 submissions for power morcellators to be marketed in the U.S. The report doesn’t provide value judgments or recommendations, but the evidence collected by the GAO team attributes the harm caused by power morcellators to:
An anemic adverse events reporting system at FDA,
A medical device clearance process that doesn’t reliably and consistently assess risk for potentially harmful devices, and
An underestimation of the risk of sarcomas occurring in uterine tissue, despite acknowledgement on FDA’s part that agency officials were “aware of the potential for spreading tissue during procedures that involved the use of power morcellators … since the agency cleared the first device in 1991.
The report is posted here.
Power morcellators, surgical devices with spinning blades, were used to perform hysterectomies and myomectomies on at least 100,000 women a year in the United States. The “minimally invasive” procedure is now known to disseminate unsuspected uterine cancer, thereby worsening a patient’s prognosis.
The report was requested by 12 members of Congress, who asked the GAO to provide a root cause analysis for harm related to morcellation, “In light of these concerns, we respectfully request you investigate the root cause failure that ultimately led to the FDA’s black box warning on the use of laparoscopic power morcellators,” legislators wrote in an Aug. 7, 2015 letter to the GAO.
The problem came to light as a result of an aggressive campaign by physicians Amy Reed and Hooman Noorchashm—along with extensive media coverage—prompted FDA to review the procedure and severely limit the use of power morcellation in November 2014.
“The GAO report confirmed the fact that literally hundreds, if not thousands, of unsuspecting American women have been harmed or died unjustifiably because of cancer upstaging by power morcellators for over two decades,” Noorchashm said to The Cancer Letter. “And it confirms that the adverse outcome reporting requirements for physicians is dangerously inadequate in the case of medical devices.”
Describing the report as “long-awaited,” Rep. Brian Fitzpatrick (R-PA) and Rep. Louise Slaughter (D-NY) said the GAO’s findings confirmed regulatory lapses at FDA that had been brought to public attention as a result of the controversy.
Fitzpatrick’s brother, Mike Fitzpatrick—whom Brian succeeded in the recent election—and Slaughter introduced in 2016 the Medical Device Guardians Act, which would require individual practitioners to report adverse events. The bill was not taken up in the 21st Century Cures Act, passed in December.
“The release of this long-awaited report won’t do anything to help women battling cancer who have had their lives devastated by power morcellators, or provide much comfort to the families of those already lost,” Brian Fitzpatrick and Slaughter said in a joint statement Feb. 8. “It does, however, shed light on the broken system that allowed this devastation to happen and include a roadmap to address it.
“The GAO report confirms what we had long expected: there are serious gaps in the FDA’s device reporting system and that immediate Congressional action is needed to reform the process and save lives. Additionally, given the associated risks, it’s clear that this device is no longer appropriate in the treatment of uterine fibroids.
“Armed with this information, we will move forward to find bipartisan legislative solutions to address these shortcomings and ensure a system is in place that provides real, accurate information to patients, professionals and regulators.”